Help
1. flow chart
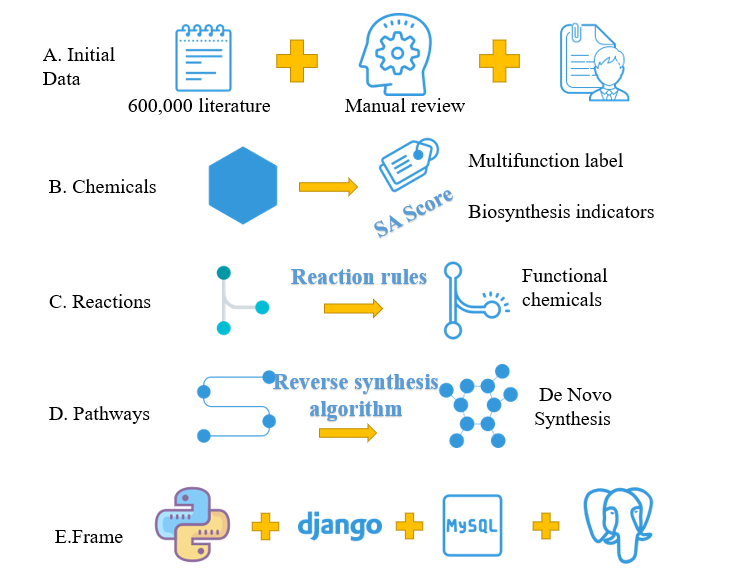
Figure.1 ChemHub flow chart
A. Constructed a data annotation system based on peer review, and manually extracted information on pathways, enzymes, and reactions from 600,000 synthetic biology literatures by experimental biologists.
B. Compares the compound information extracted from the literature with multiple functional molecule compound databases, deduplicates with SMILES, and gives the synthetic accessibility score (SA Score).
C. Collect reaction data and extract reaction rules. Tools based on reaction rules seek for precursors of functional compounds.
D. Summarize the pathway data and de novo design a synthetic pathway for functional compounds based on the reverse synthesis algorithm.
E. website framework is built with Django 1.6, the database PostgreSQL and MySQL are used together, and the front end uses HTML, CSS, jQuery for rendering.
2. overview

Figure.2 Database overview
1. The homepage of ChemHub, five search methods, compound name search, chemical structural formula search, similarity, fragmentation and maximum common substructure search. And contains two tools Biotransformation to explore biological precursors and de novo pathways design.
2. JME molecular tool is convenient for users to draw and convert molecular structure into SMILES
3. Functional classification labels of ChemHub compounds, SA score and some basic information.
4. Use ECharts to visualize the biosynthetic pathway of functional compounds.
5. Use darge to visualize the biosynthetic pathway of functional compounds.
6. Use nwet to visually display the biosynthetic pathways of functional compounds to distinguish the genetic information of subcellular structures and metabolites.
7. BCSExplorer performs biotransformation based on chemical reaction rules to find precursors of functional compounds.
3. text query
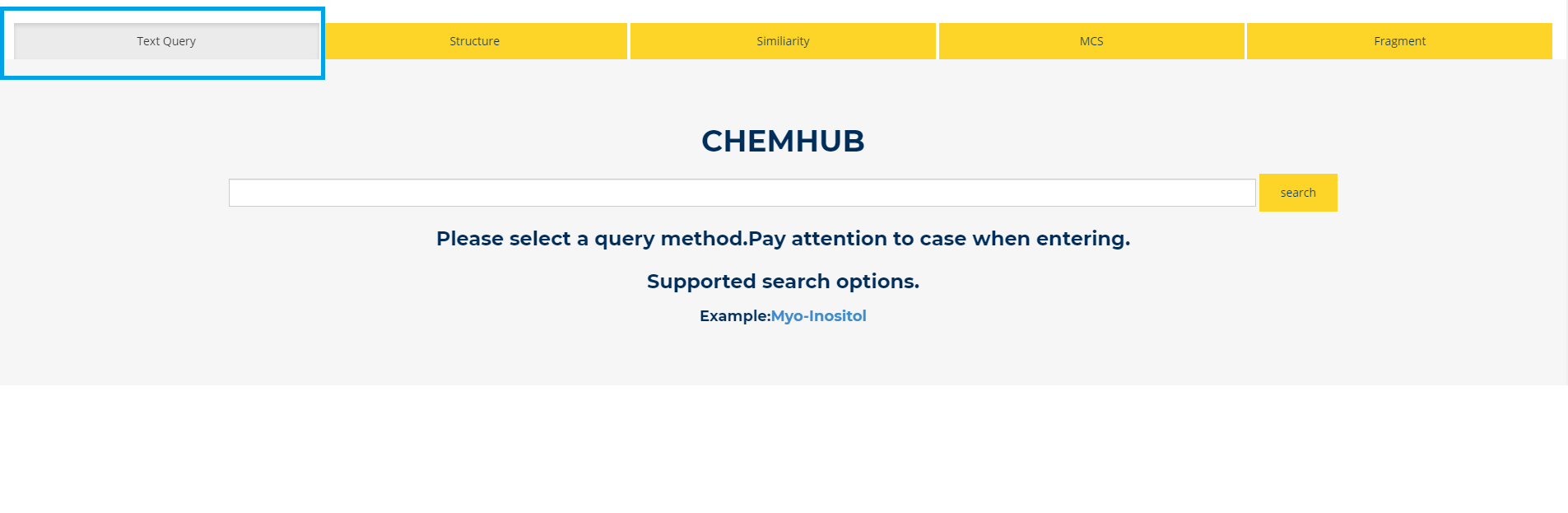
Figure.3 Text Query You can retrieve the biosynthetic compounds interested in by the name of the compound.
4. Structure
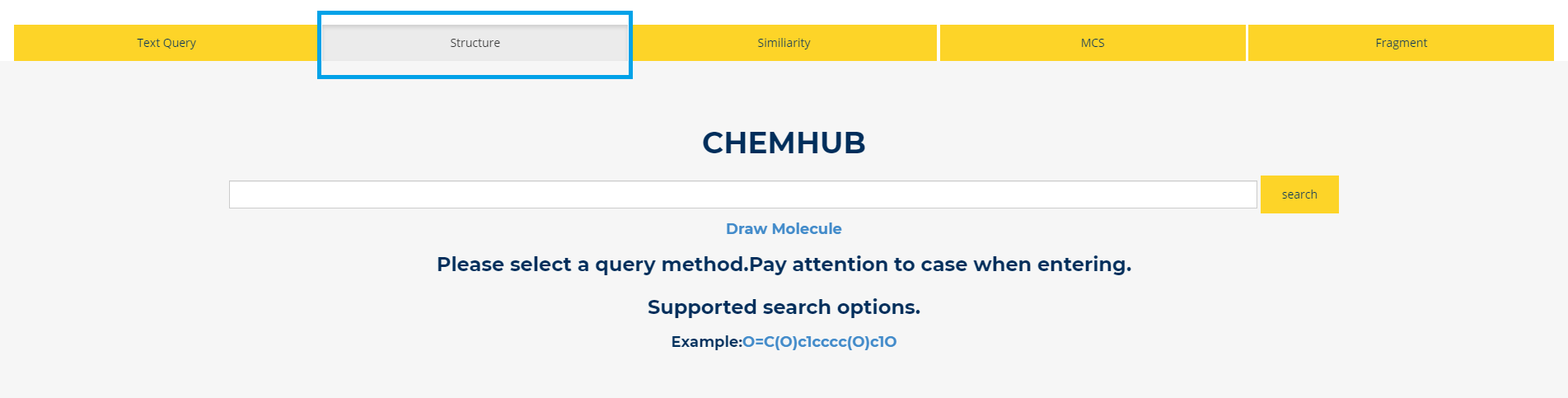
Figure.4 Structure Query You can retrieve the biosynthetic compounds interested in by the structure(SMILES) of the compound.
5. Similarity
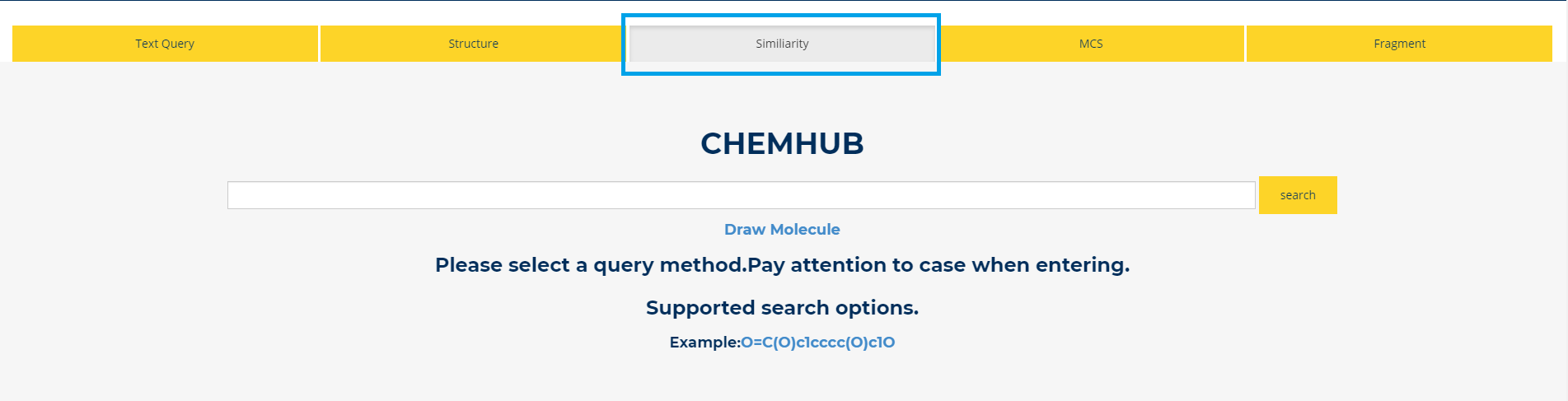
Figure.5 Similarity Query For the similarity search, we adopt Tanimoto similarity measure by comparing chemical structures represented by means of 2D fingerprints. Clicking on the Similarity option in the new page, 20 compounds with the highest similarity compared to input compound in the ChemHub library will be displayed.
6. MCS
Figure.6 MCS Query The MCS (maximum common substructure) option is proposed as a new tool to make it easy to group compounds which share the same pattern. In particular, the MCS algorithm employed in this study is the algorithm fMCS for speed. Click on the MCS option in the new page and 20 compounds with the largest common substructure compared to input compound in the ChemHub library will be displayed.
7. fragment
Figure.7 Fragment Query Fragment is defined to be made up of a set of connected with atoms that have associated functional groups. The key of this search option is in essence a graph comparison algorithm. Click on the new page of Fragment options, 20 compounds with a common fragment compared to input compound in ChemHub library will be displayed.
8. Exploring biosynthetic precursors
Figure.8 Exploring biosynthetic precursors Enter the SMILES of the compound of interest in the input box, select the enzyme type, atomic ratio, and similarity to return to the precursors of the current compound under analysis, druggability analysis and toxicity analysis.
(1) SMILES: the query compound can be represented as SMILES input by the user or drawn with a molecular input editor.
(2) Transformations: the user could specify transformation(s) of reactions which would be used to explore precursors.
(3) Atoms ratio: all products with atoms ratio higher than specified would be discarded.
(4) Similarity: all transformations with reactant similarity higher than specified would be used to explore precursors.
(5) Analysis: the user could specify analysis of compounds which would be used to rank all possible compounds.
Results
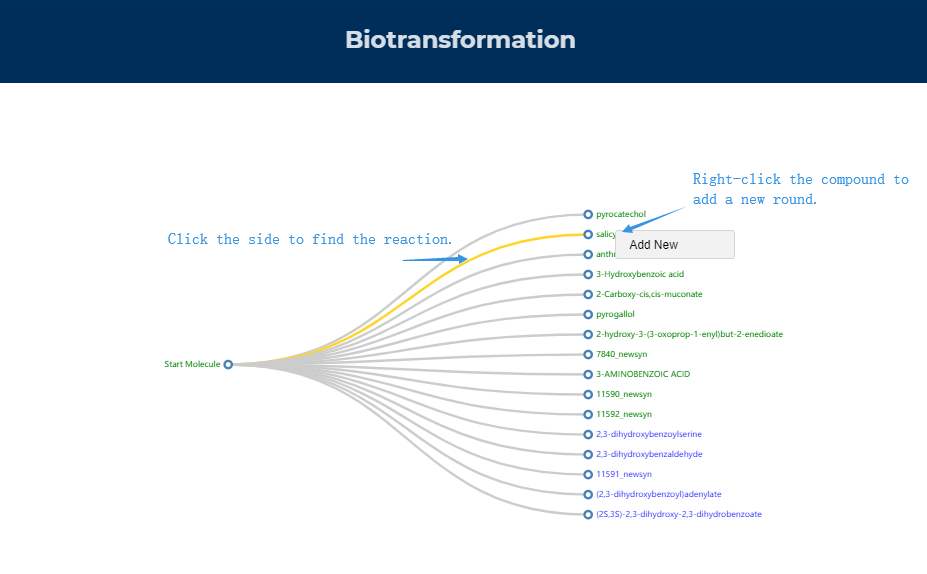
Figure.9 The results of precursor exploration are displayed.
9. The animation of the biotransformation
Figure.10 The use of biotransformation animation display.
10. De novo pathways design
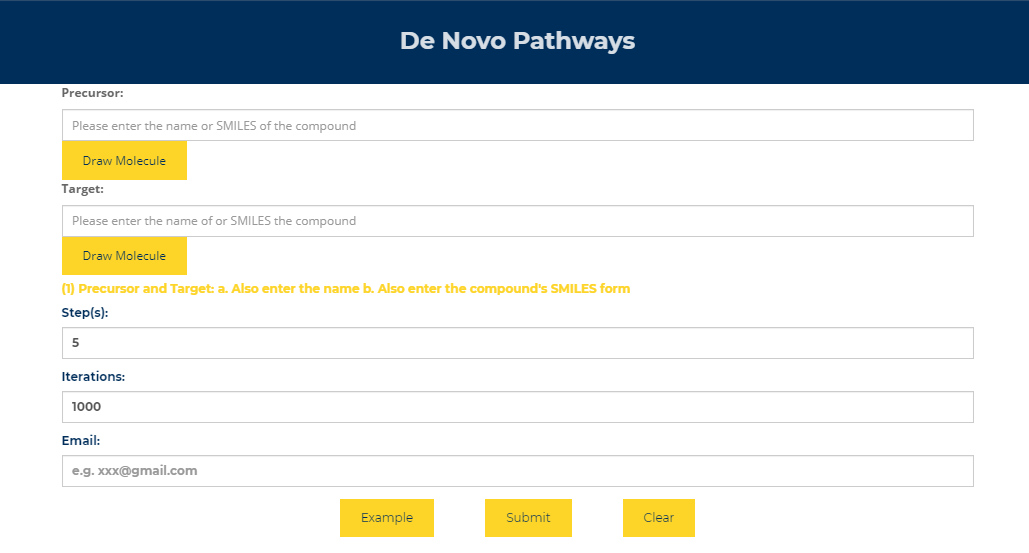
Figure.11 De novo pathways design Enter the name of the precursor compound of interest and the target compound, enter the maximum path length and number of iterations, and enter your Email. Subsequent results will be sent to your mailbox, or you can choose to stay in this interface and wait for the results to return. The larger the path length and the number of iterations, the longer the calculation time.
(1) Precursor and Target: a. Also enter the name b. Also enter the compound's SMILES form
(2) Step(s):It must be an integer, which means that how many reactions involved at most. Of course, the computing time will be prolonged with its increasment,the maximum number of reactions is limited to 10 for the sake of computation time.
(3) Iterations:Because the retrosynthesis algorithm utilized in ChemHub is iterative, users should enter the iteration-searching times. The retrosynthesis algorithm proceeds until the customized iterations are reached, and each iteration will stop until the predefined steps or the precursor/sink metabolites are reached. For example: 1,000 times or more. Of course, the computation time will be prolonged with its increasment.
no result
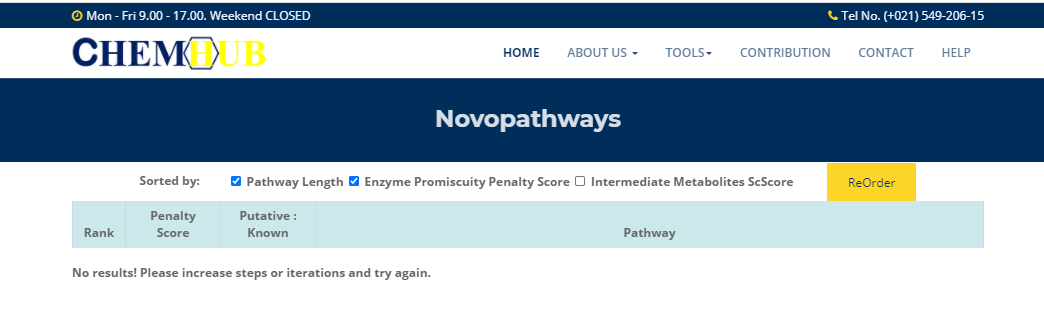
Figure.12 No results!
If the result shown in the figure appears, it is a normal phenomenon. Please increase steps or iterations and try again.
11. The animation of the De novo (1)
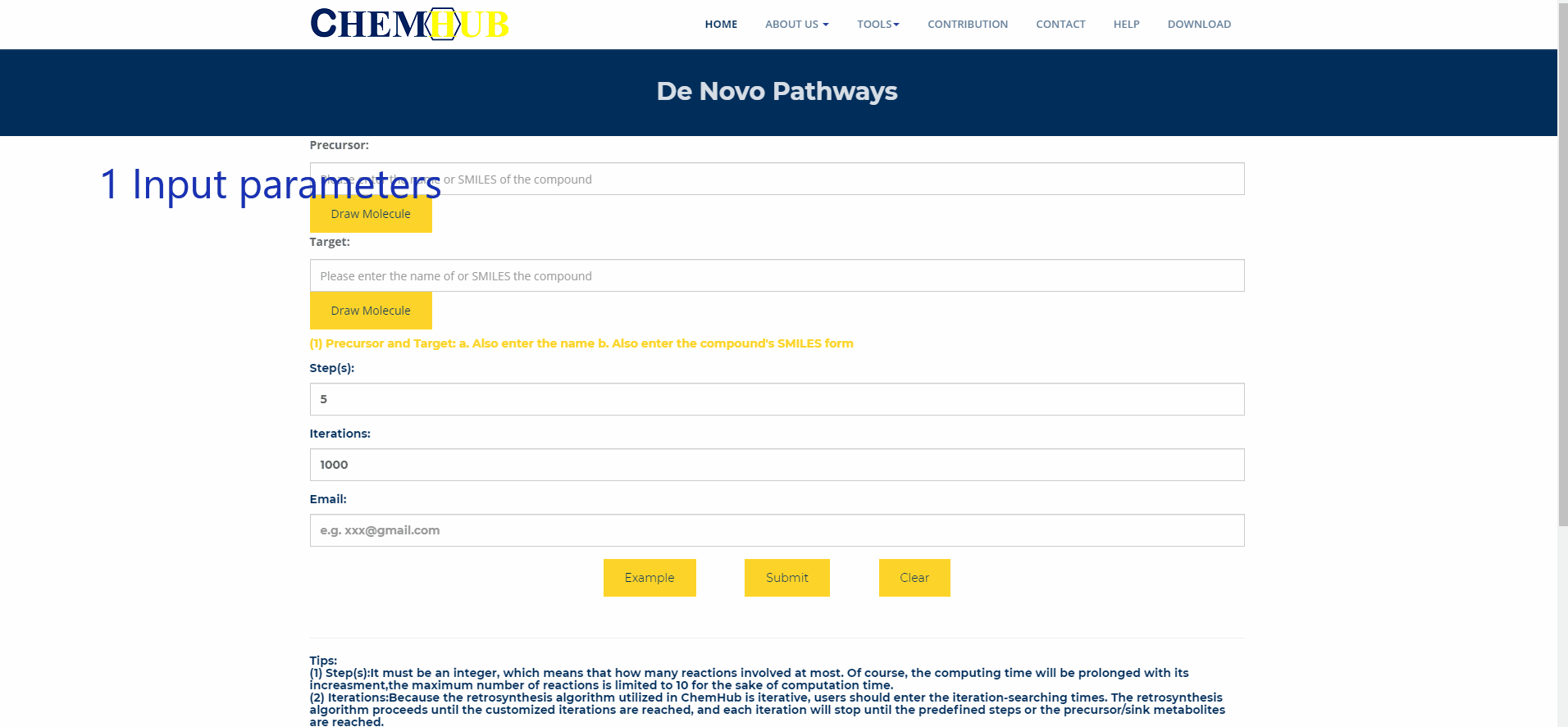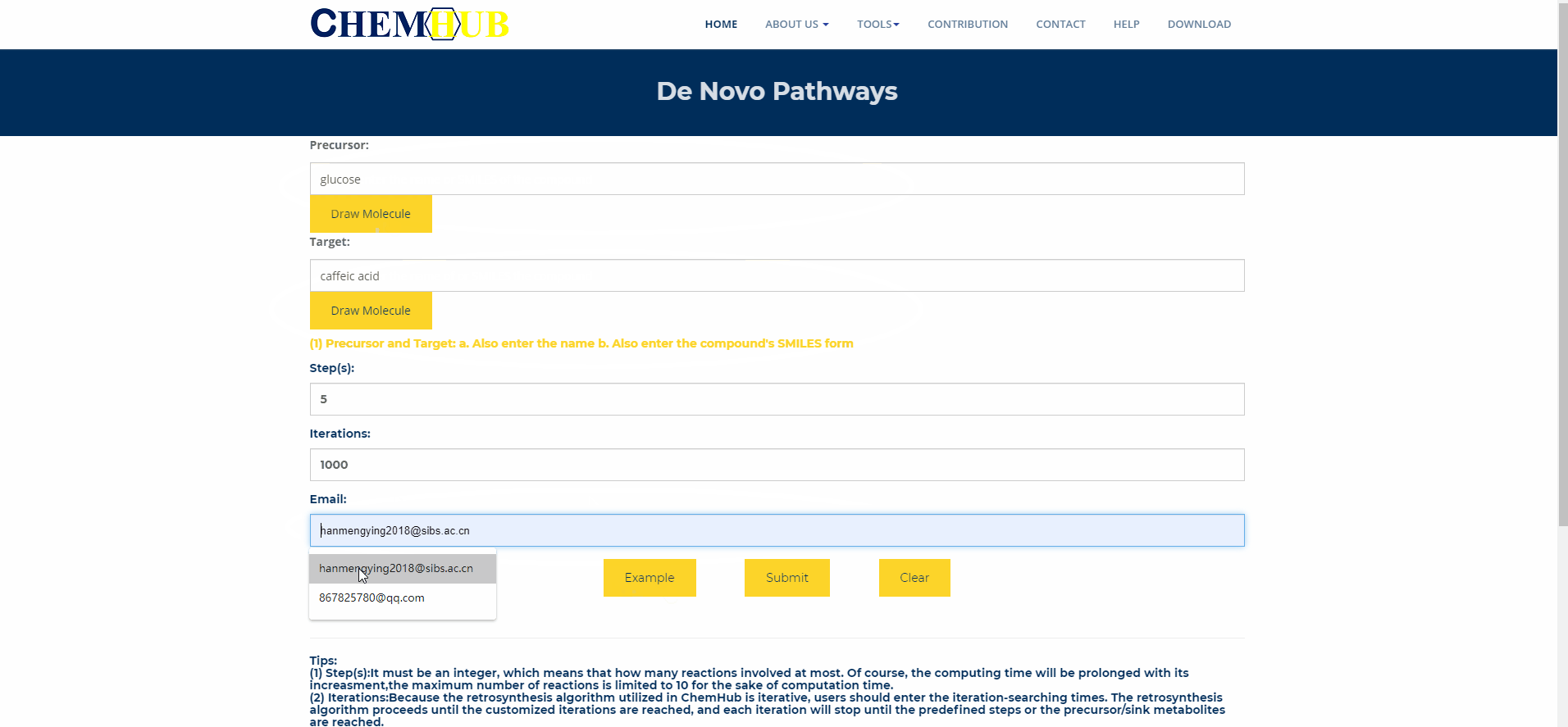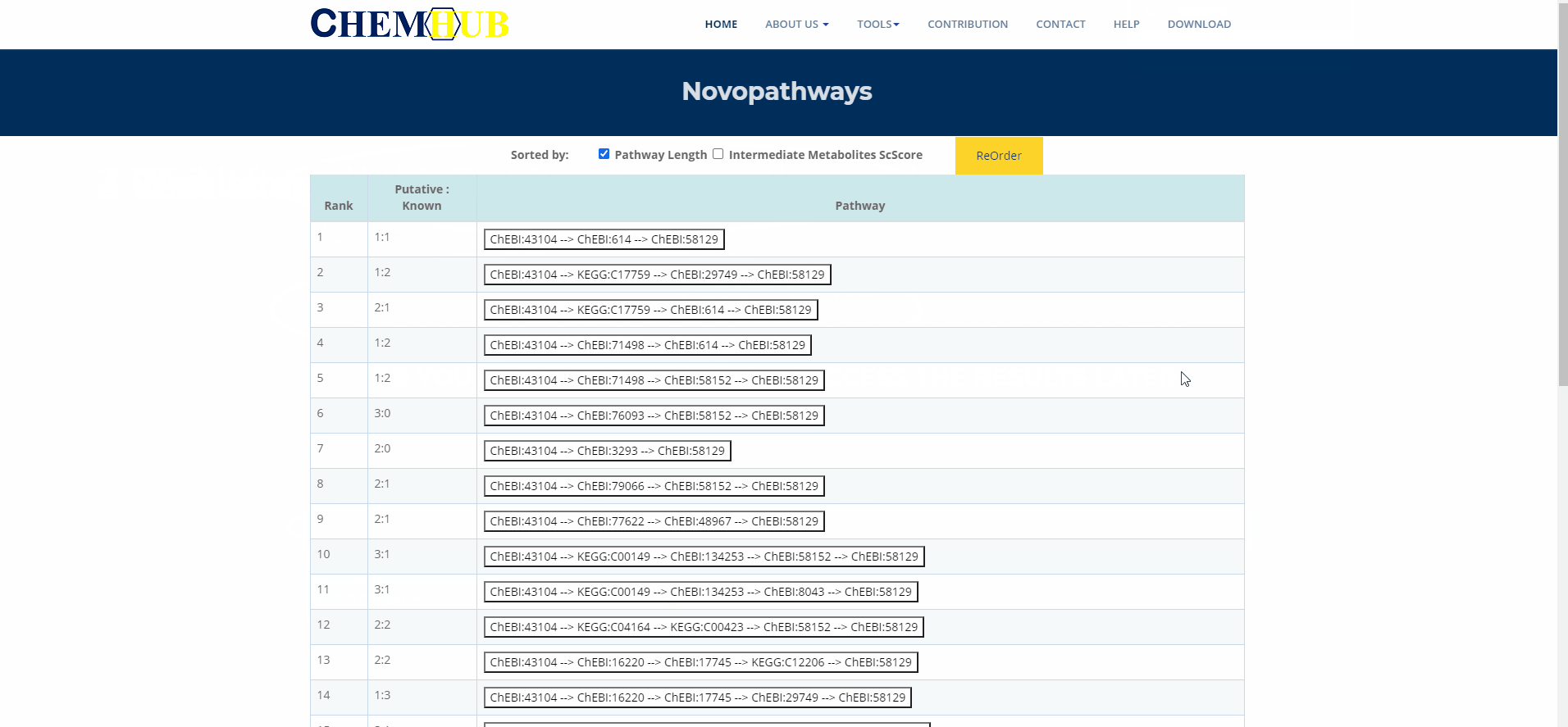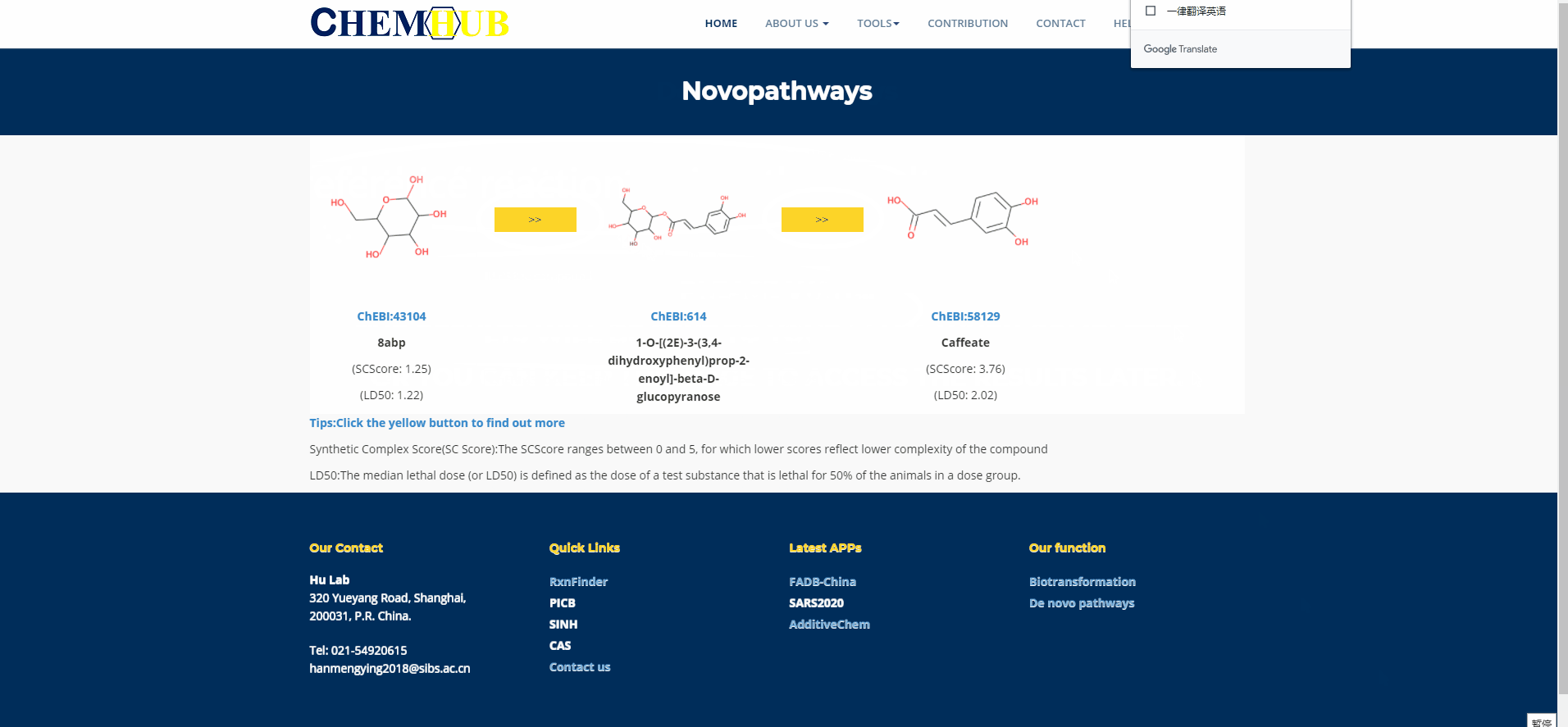
Figure.13 The use of denovo animation display(1).
12. The animation of the De novo (2)

Figure.14 The use of denovo animation display(2).
13. The animation of the De novo (3)
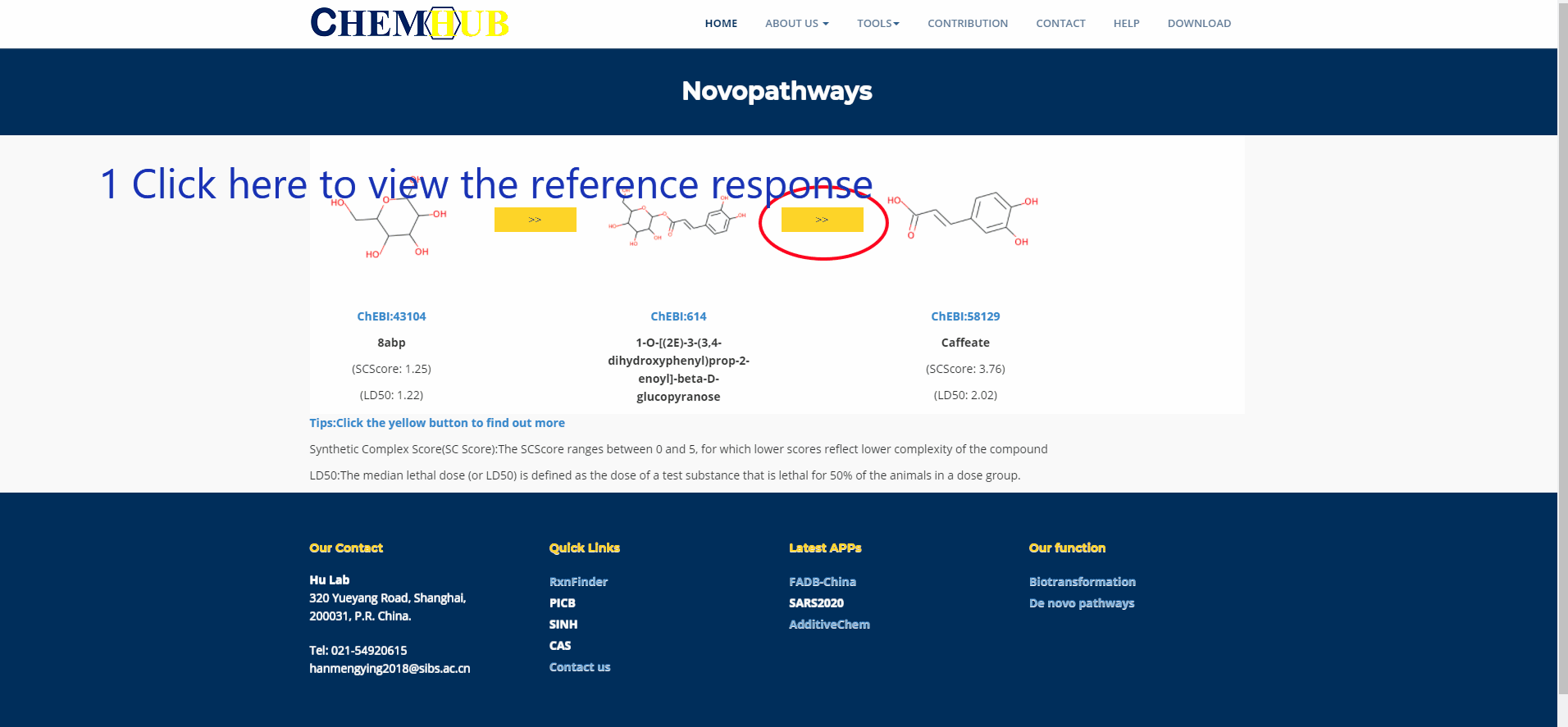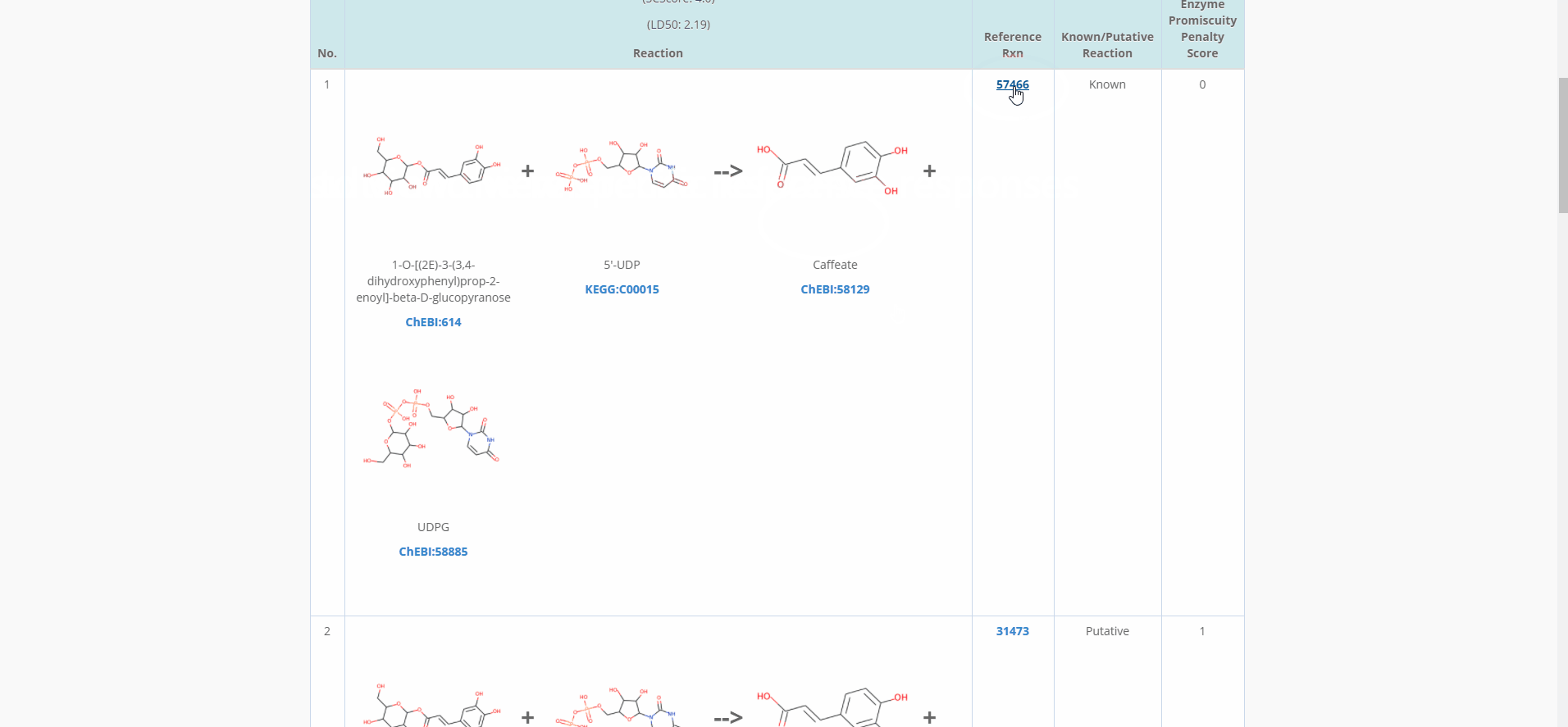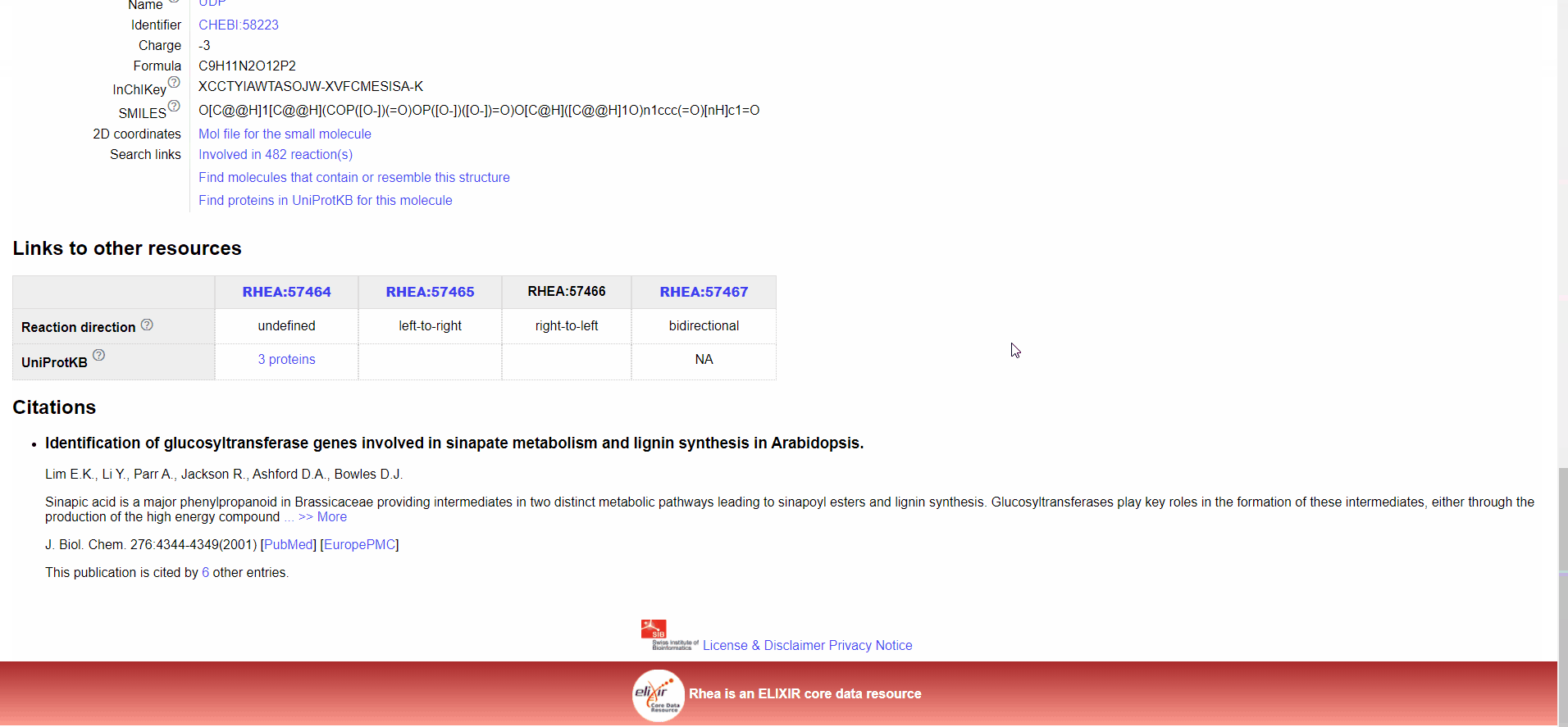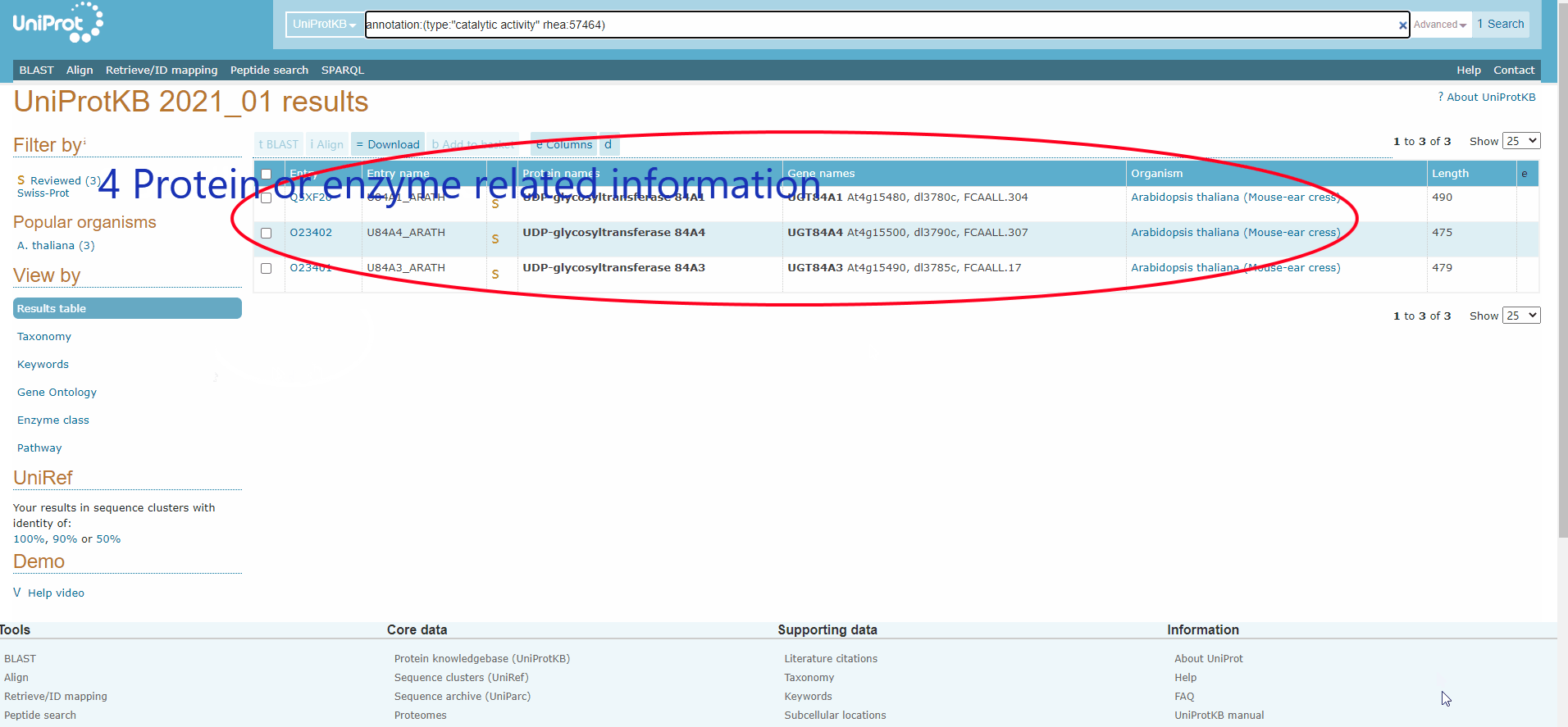
Figure.15 The use of denovo animation display(3).